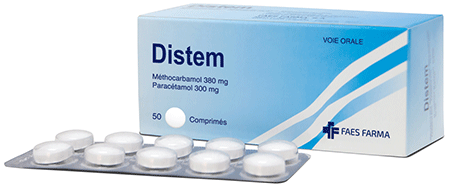
Distem® comprimé
1 – DÉNOMINATION DU MEDICAMENT
Distem® 380 mg/300 mg comprimés
2 – COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé contient 380 mg de méthocarbamol et 300 mg de paracétamol.
Pour la liste complète des excipients, voir rubrique 6.1.
3 – FORME PHARMACEUTIQUE
Comprimés.
Les comprimés sont blancs, ronds, biconvexes et comportent une barre de cassure sur une face.
La barre de cassure permet seulement de faciliter la prise du comprimé, elle ne le divise pas en doses égales.
4 – DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Traitement symptomatique à court terme des spasmes musculaires douloureux liés aux troubles musculosquelettiques aigus.
4.2 Posologie et mode d’administration
Posologie
Le traitement par le méthocarbamol doit être le plus court possible. Lorsque les symptômes douloureux disparaissent, l’administration du médicament doit être interrompue.
Adultes : 2 comprimes toutes les 4-6 heures (soit quatre à six fois par jour), en fonction de l’intensité des symptômes. Ne pas dépasser 12 comprimes par 24 heures.
Patients souffrant d’une insuffisance rénale
En cas d’insuffisance rénale, il convient de réduire la dose en fonction de la valeur du débit de filtration glomérulaire selon le tableau suivant :
| Filtration glomérulaire | Dose |
| 10 à 50 ml/min | 1 comprimé toutes les 6 heures |
| < 10 ml/min | 1 comprimé toutes les 8 heures |
Patients souffrant d’une insuffisance hépatique
En cas d’insuffisance hépatique, il convient de ne pas dépasser 2 grammes de paracétamol par 24 heures (soit 6 comprimés par jour au maximum, répartis en 3-6 prises par jour) [voir rubrique 4.4].
Compte tenu du fait que la demi-vie d’élimination du méthocarbamol peut être plus longue chez ces patients, un intervalle plus long entre les prises des comprimés peut s’avérer nécessaire.
Patients âgés
Chez les patients âgés, une augmentation de la demi-vie d’élimination du paracétamol a été observée et il est apparu que la moitié de la dose de méthocarbamol peut être suffisante pour obtenir une réponse thérapeutique. Il est donc recommandé de réduire la dose à 1 comprimé toutes les 4 heures ou à 2 comprimés toutes les 8 heures.
Mode d’administration
L’ingestion des comprimes peut être facilitée par une petite quantité d’eau.
4.3 Contre-indications
- Hypersensibilité connue aux substances actives (méthocarbamol, paracétamol) ou à l’un des excipients mentionnés à la rubrique 6.1
- État comateux ou pré-comateux.
- Pathologie cérébrale connue.
- Antécédents de crises convulsives ou d’épilepsie.
- Myasthénie grave
4.4 Mises en garde spéciales et précautions d’emploi
L’utilisation simultanée de plus d’un médicament contenant du paracétamol peut provoquer des tableaux d’intoxication (voir rubrique 4.9). Le patient doit être averti du risque éventuel d’intoxication lié à l’utilisation simultanée accidentelle de plus d’un médicament contenant du paracétamol. Les tableaux toxiques liés au paracétamol peuvent survenir suite à l’ingestion d’une dose excessive unique ou à l’accumulation résultant de prises répétées de doses excessives de paracétamol (voir rubrique 4.9).
Ce médicament doit être administré avec prudence, en évitant les traitements prolongés chez les patients souffrant d’un déficit en glucose-6-phosphate déshydrogénase, d’une anémie, d’affections cardiaques ou pulmonaires.
L’utilisation du médicament chez les patients consommant régulièrement de l’alcool (trois boissons alcoolisées ou plus par jour) peut provoquer des lésions hépatiques. Chez les alcooliques chroniques, il convient de ne pas administrer plus de 2 g de paracétamol par jour (soit 5 comprimés par jour au maximum). La consommation d’alcool est déconseillée pendant le traitement par le paracétamol.
La prudence est de mise chez les patients asthmatiques sensibles à l’acide acétylsalicylique car l’apparition de légères réactions bronchospastiques a été rapportée avec le paracétamol (réaction croisée) chez ces patients (voir rubrique 4.8) ; bien qu’elles ne se soient manifestées que chez une minorité de ces patients, le paracétamol peut provoquer des réactions graves dans certains cas, surtout lorsqu’il est administré à des doses élevées.
Populations particulières
Insuffisance rénale et hépatique
Ce médicament doit être utilisé avec prudence chez les patients souffrant d’une insuffisance rénale ou hépatique.
Chez les patients souffrant d’une insuffisance hépatique et rénale, le méthocarbamol doit être administré avec prudence, en évitant les traitements prolongés.
Les patients souffrant d’une insuffisance rénale doivent consulter le médecin ou le pharmacien avant de prendre le médicament car un ajustement de la dose peut s’avérer nécessaire (voir rubrique 4.2). En cas d’insuffisance rénale grave, l’utilisation occasionnelle de paracétamol est tolérée, mais l’administration prolongée de doses élevées peut augmenter le risque d’apparition d’effets hépatiques et rénaux indésirables.
La prudence est recommandée lors de l’administration de paracétamol chez les patients présentant une insuffisance hépatique légère à modérée (incluant le syndrome de Gilbert), une insuffisance hépatique grave (Child-Pugh > 9), une hépatite aiguë, un déficit en glutathion, une déshydratation, une malnutrition chronique et des antécédents d’abus d’alcool (voir rubrique 4.2), ainsi que lors du traitement simultané avec des médicaments inducteurs des enzymes hépatiques (voir rubrique 4.5).
Interférences avec les tests de diagnostic
Le paracétamol peut modifier les valeurs des déterminations analytiques de l’acide urique et du glucose.
Le méthocarbamol peut interférer avec la couleur de certains tests analytiques tels que la détermination de l’acide 5-hydroxy-indolacétique (5-HIAA) utilisant le nitrosonaphtol comme réactif et la détermination de l’acide vanillylmandélique (AVM) par la méthode Gitlow. Il a également été rapporté une altération de la couleur des échantillons d’urine de certains patients pendant le stockage, qui devient marron, noire, bleue ou verte.
4.5 Interactions avec d’autres médicaments et autres formes d’interactions
Méthocarbamol
Le méthocarbamol peut augmenter les effets d’autres agents dépresseurs et stimulants du système nerveux central, y compris l’alcool, les barbituriques, les anesthésiques et les coupe-faim.
Il peut également renforcer les effets des anticholinergiques tels que l’atropine et certains médicaments psychotropes.
Le méthocarbamol peut inhiber l’effet du bromure de pyridostigmine. Par conséquent, il doit être utilisé avec prudence chez les patients souffrant d’une myasthénie grave traités par des inhibiteurs de l’acétylcholinestérase.
Paracétamol
Le paracétamol est métabolisé au niveau hépatique, entraînant la formation de métabolites hépatotoxiques. Il peut donc interagir avec des médicaments utilisant ses mêmes voies de métabolisation. Ces médicaments sont les suivants :
- Anticoagulants oraux (acénocoumarol, warfarine) : l’administration chronique de doses de paracétamol supérieures à 2 g/jour conjointement à ce type de médicaments peut provoquer une augmentation de l’effet anticoagulant, probablement en raison de la réduction, par interférence avec les enzymes impliquées dans la synthèse hépatique, des facteurs de la coagulation dépendants de la vitamine K. Les interactions entre le paracétamol et les anticoagulants oraux peuvent renforcer l’effet anticoagulant, ce qui entraîne un risque d’hémorragies. Compte tenu de la faible pertinence clinique apparente à des doses inférieures à 2 g/jour, une alternative thérapeutique par l’administration de salicylés doit être envisagée chez les patients recevant un traitement anticoagulant. Toutefois, il est recommandé de surveiller régulièrement l’INR chez ces patients.
- Alcool éthylique : potentialisation de la toxicité du paracétamol par une induction possible de la production hépatique de substances hépatotoxiques dérivées du paracétamol.
- Anticholinergiques (glycopyrronium, propanthéline) : diminution de l’absorption du paracétamol avec une inhibition possible de son effet en raison de la réduction de la vitesse de la vidange gastrique.
- Anticonvulsivants (phénytoïne, phénobarbital, méthylphénobarbital, primidone) : réduction de la biodisponibilité du paracétamol et potentialisation de l’hépatotoxicité en cas de surdosage en raison de l’induction du métabolisme hépatique.
- Diurétiques de l’anse : les effets des diurétiques peuvent être réduits car le paracétamol peut réduire l’excrétion rénale des prostaglandines et l’activité de la rénine plasmatique.
- Isoniazide : diminution de la clairance du paracétamol, avec potentialisation possible de son action et/ou de sa toxicité, par inhibition de son métabolisme hépatique.
- Lamotrigine : diminution de la biodisponibilité de la lamotrigine, avec réduction possible de son effet, due à l’induction possible de son métabolisme hépatique.
- Métoclopramide et dompéridone : augmentent l’absorption du paracétamol dans l’intestin grêle en raison de leur effet sur la vidange gastrique, et donc le début d’action.
- Probénécide : augmente la demi-vie plasmatique du paracétamol en réduisant la dégradation et l’excrétion urinaire de ses métabolites.
- Propranolol : augmente les taux plasmatiques de paracétamol par une éventuelle inhibition de son métabolisme hépatique.
- Rifampicine : augmente la clairance du paracétamol et la formation de ses métabolites hépatotoxiques en raison d’une induction possible de son métabolisme hépatique.
- Résines échangeuses d’ions (cholestyramine) : diminution de l’absorption du paracétamol, avec une inhibition possible de son effet, due à la fixation du paracétamol dans l’intestin.
- Chloramphénicol : potentialisation de la toxicité du chloramphénicol par une inhibition possible de son métabolisme hépatique.
- Zidovudine (AZT) : son utilisation simultanée avec le paracétamol augmente le risque de réduire le taux de globules blancs (neutropénie). Par conséquent, le paracétamol ne doit pas être administré avec l’AZT, sauf en cas d’indication médicale.
4.6 Fertilité, grossesse et allaitement
4.6.1 Grossesse
Le paracétamol traverse la barrière placentaire.
Aucune étude sur la reproduction chez les animaux ayant reçu du méthocarbamol n’a été réalisée. On ignore si le méthocarbamol peut provoquer des dommages fœtaux ou influencer la capacité reproductive lorsqu’il est administré à des femmes enceintes.
La sécurité d’utilisation du méthocarbamol concernant les effets indésirables éventuels sur le développement fœtal n’a pas été établie. Il existe des rapports isolés faisant état d’anomalies fœtales et congénitales après une exposition utérine au méthocarbamol.
L’utilisation de ce médicament chez les femmes enceintes ou susceptibles de l’être n’est pas recommandée, en particulier pendant les phases initiales de la grossesse, à moins que le médecin ne considère que les bénéfices potentiels sont supérieurs aux éventuels risques d’utilisation.
4.6.2 Allaitement
Chez la chienne, le méthocarbamol et/ou ses métabolites sont excrétés dans le lait maternel. On ignore toutefois si c’est également le cas chez la femme.
Le paracétamol est excrété dans le lait maternel mais en quantités non cliniquement significatives.
Il faudra donc être prudent lors de l’administration de Distem chez la femme allaitante.
4.6.3 Fertilité
Fertilité masculine (voir rubrique 5.3).
4.7 Effet sur l’aptitude à la conduite et l’utilisation de machines
Ce médicament peut provoquer une somnolence. Les patients ne doivent donc pas conduire de véhicules ni utiliser de machines, à moins que leurs capacités mentales paraissent inaltérées, en particulier si d’autres médicaments pouvant entraîner une somnolence sont administrés simultanément.
4.8 Effets indésirables
Effets indésirables rapportés liés au méthocarbamol
Il est impossible de déterminer l’estimation des fréquences des effets indésirables liés à l’administration de méthocarbamol. Les effets indésirables rapportés après l’administration de méthocarbamol sont les suivants :
Affections hématologiques et du système lymphatique : leucopénie.
Affections du système nerveux : nervosité, fatigue, anxiété, tremblement, amnésie, confusion, étourdissements, vertige, somnolence, insomnie, légère incoordination musculaire, convulsions (incluant le grand mal).
Affections cutanées et altérations particulières des sens : diplopie, vision trouble, conjonctivite accompagnée de congestion nasale, nystagmus, goût métallique, prurit, éruption cutanée, urticaire.
Affections cardiaques : bradycardie, rougeur, hypotension, syncope.
Affections gastro-intestinales : dyspepsie, nausées et vomissements, dysgueusie.
Affections hépatobiliaires : ictère (incluant l’ictère cholestatique).
Troubles généraux : œdème angioneurotique, réaction anaphylactique, fièvre, maux de tête.
Affections de la peau et du tissu sous-cutané : prurit, éruption cutanée, urticaire.
Effets indésirables rapportés liés au paracétamol
En général, les effets indésirables liés au paracétamol sont rares (fréquence ≥ 1/10 000, < 1/1 000) ou très rares (fréquence < 1/10 000) et habituellement d’intensité légère. Les effets indésirables les plus fréquemment rapportés au cours de l’utilisation du paracétamol sont les suivants : hépatotoxicité, toxicité rénale, anomalies de la formule sanguine, hypoglycémie et dermatite allergique.
Affections hématologiques et du système lymphatique
Très rare : thrombocytopénie, agranulocytose, leucopénie, neutropénie, pancytopénie, anémie hémolytique.
Troubles du métabolisme et de la nutrition
Très rare : hypoglycémie.
Affections cardiaques
Rare : hypotension.
Affections hépatobiliaires
Rare : élévation des taux de transaminases hépatiques.
Très rare : hépatotoxicité (ictère).
Affections respiratoires, thoraciques et médiastinales
Très rare : bronchospasme.
Affections de la peau et du tissu sous-cutané
Très rare : dermatite allergique.
Dans de très rares cas, des réactions cutanées graves, incluant le syndrome de Stevens-Johnson (SSJ) et la nécrolyse épidermique toxique, ont été rapportées.
Affections du rein et des voies urinaires
Très rare : pyurie stérile (urine trouble), effets rénaux indésirables surtout en cas de surdosage (voir rubrique 4.4).
Troubles généraux
Rare : malaise.
Très rare : réactions d’hypersensibilité allant d’une simple éruption cutanée (rash) à de l’urticaire, à un angio-œdème et au choc anaphylactique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable via le Système National de Pharmacovigilance du pays.
4.9 Surdosage
Méthocarbamol
Il existe peu d’informations sur la toxicité aiguë du méthocarbamol. Les cas de surdosage par le méthocarbamol sont associés à l’ingestion d’alcool et à d’autres agents dépresseurs du SNC. Les symptômes suivants ont été observés : nausées, étourdissements, vision trouble, hypotension, convulsions et coma. Des cas particuliers d’ingestion de quantités supérieures à 22-30 grammes de méthocarbamol sans toxicité grave et de survie/récupération après l’ingestion de 30-50 grammes ont été rapportés. Dans les deux cas, le symptôme principal était une somnolence extrême. Le traitement a été symptomatique et dans les deux cas la récupération s’est produite sans autres effets. Cependant, des cas de surdosage d’évolution fatale ont été rapportés.
Le traitement du surdosage par le méthocarbamol comporte un traitement symptomatique et de soutien. Les mesures de soutien comprennent le maintien d’une voie aérienne, la surveillance de l’excrétion urinaire et des signes vitaux et, si nécessaire, l’administration de fluides intraveineux. L’utilité de l’hémodialyse en cas de surdosage n’est pas connue.
Paracétamol
Dans le cas du paracétamol, la symptomatologie liée à un surdosage comprend nausées, vomissements, perte d’appétit, ictère, douleur abdominale et insuffisances rénale et hépatique. En cas d’ingestion d’une dose excessive, le patient doit être rapidement traité dans un centre médical, même en l’absence de symptômes significatifs, ceux-ci ne se manifestant en général pas immédiatement après l’ingestion, mais seulement à partir du troisième jour. La mort peut survenir par nécrose hépatique. Une insuffisance rénale aiguë peut également se développer.
Le surdosage par le paracétamol est évalué en quatre phases, qui commencent au moment de l’ingestion de la dose excessive :
- PHASE I (12 à 24 heures) : nausées, vomissements, diaphorèse et anorexie.
- PHASE II (24 à 48 heures) : amélioration clinique ; les taux d’AST, d’ALT, de bilirubine et de prothrombine commencent à augmenter.
- PHASE III (72 à 96 heures) : pic d’hépatotoxicité ; l’AST peut atteindre des valeurs de 20 000.
- PHASE IV (7 à 8 jours) : récupération.
Une hépatotoxicité peut se manifester. La dose toxique minimale est de 6 g chez l’adulte et de plus de 100 mg/kg de poids chez l’enfant. Des doses supérieures à 20-25 g sont potentiellement mortelles. Les symptômes de l’hépatotoxicité comprennent nausées, vomissements, anorexie, mal-être, diaphorèse, douleurs abdominales et diarrhée. L’hépatotoxicité se manifeste seulement 48 à 72 heures après l’ingestion. Si la dose ingérée est supérieure à 150 mg/kg ou si elle ne peut être déterminée, il faut prélever un échantillon de paracétamol sérique 4 heures après l’ingestion. En cas d’hépatotoxicité, un examen de la fonction hépatique doit être réalisé et répété toutes les 24 heures. L’insuffisance hépatique peut entraîner une encéphalopathie, le coma et la mort.
Les concentrations plasmatiques de paracétamol supérieures à 300 microgrammes/ml détectées 4 heures après l’ingestion ont été associées à des lésions hépatiques chez 90 % des patients. Celles-ci commencent à se manifester lorsque les taux plasmatiques de paracétamol sont inférieurs à 120 microgrammes/ml au bout de 4 heures ou inférieurs à 30 microgrammes/ml au bout de 12 heures.
L’ingestion chronique de doses supérieures à 4 g/jour peut donner lieu à une hépatotoxicité transitoire. Les reins peuvent subir une nécrose tubulaire et le myocarde peut être lésé.
Traitement : dans tous les cas, il faudra procéder à une aspiration et à un lavage gastrique, de préférence dans les 4 heures suivant l’ingestion.
Il existe un antidote spécifique pour la toxicité produite par le paracétamol : la N-acétylcystéine. Chez les patients adultes, on recommande l’administration de 300 mg/kg de N-acétylcystéine (équivalant à 1,5 ml/kg de solution aqueuse à 20 % ; pH : 6,5), par voie IV pendant une période de 20 heures et 15 minutes, conformément au schéma suivant :
- Dose d’attaque : 150 mg/kg (équivalant à 0,75 ml/kg de solution aqueuse à 20 % de N-acétylcystéine ; pH : 6,5) par voie intraveineuse lente ou dilués dans 200 ml de dextrose à 5 %, pendant 15 minutes.
- Dose d’entretien :
a) Administration initiale de 50 mg/kg (équivalant à 0,25 ml/kg de solution aqueuse à 20 % de N-acétylcystéine ; pH: 6,5) dans 500 ml de dextrose à 5 % en perfusion lente pendant 4 heures.
b) Puis administration de 100 mg/kg (équivalant à 0,50 ml/kg de solution aqueuse à 20 % de N-acétylcystéine ; pH : 6,5) dans 1 000 ml de dextrose à 5 % en perfusion lente pendant 16 heures.
L’efficacité de l’antidote est maximale s’il est administré dans les 8 heures suivant l’intoxication. L’efficacité diminue progressivement à partir de la huitième heure, et elle est nulle 15 heures après l’intoxication.
L’administration de la solution aqueuse de N-acétylcystéine à 20 % peut être interrompue lorsque les résultats de l’examen sanguin affichent des concentrations sanguines de paracétamol inférieures à 200 microgrammes/ml.
Effets indésirables liés à l’administration de la N-acétylcystéine par voie IV
Des éruptions cutanées et une réaction anaphylactique ont exceptionnellement été observées, généralement entre 15 minutes et 1 heure après le début de la perfusion.
Administré par voie orale, l’antidote N-acétylcystéine doit être pris dans les 10 heures suivant le surdosage. La dose d’antidote recommandée chez l’adulte est la suivante :
– une dose initiale de 140 mg/kg de poids corporel
– 17 doses de 70 mg/kg de poids corporel, soit une prise toutes les 4 heures
Chaque dose doit être diluée à 5 % dans un soda, un jus de raisin ou d’orange ou de l’eau, avant d’être administrée, à cause de son odeur désagréable et de ses propriétés irritantes ou sclérosantes. Si la dose est vomie dans l’heure qui suit l’administration, elle doit être répétée. Le cas échéant, l’antidote (dilué dans de l’eau) peut être administré par intubation duodénale.
5 – PROPRIÉTÉS PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : myorelaxants, esters de l’acide carbamique, méthocarbamol en association, sauf avec psycholeptiques.
Code ATC : M03BA53
Méthocarbamol
Le méthocarbamol est un myorelaxant à action centrale. Il provoque son effet myorelaxant par inhibition de la conduction réflexe polysynaptique au niveau de la moelle épinière et des centres sous-corticaux. Aux doses thérapeutiques, il n’affecte ni le tonus physiologique ni la contractilité de la musculature squelettique ni la motilité de la musculature lisse.
Paracétamol
Le paracétamol est un médicament analgésique qui possède également des propriétés antipyrétiques.
Le mécanisme d’action exact du paracétamol est inconnu, bien que l’on sache qu’il agit au niveau du système nerveux central et, dans une moindre mesure, par blocage de la génération de l’influx douloureux au niveau périphérique. On pense que le paracétamol relève le seuil de la douleur en inhibant la synthèse des prostaglandines par blocage des cyclo-oxygénases dans le système nerveux central (en particulier la COX-3). Néanmoins, le paracétamol n’inhibe pas de manière significative les cyclo-oxygénases des tissus périphériques.
Le paracétamol stimule l’activité des voies sérotoninergiques descendantes qui bloquent la transmission vers la moelle épinière des signaux nociceptifs provenant des tissus périphériques. Certaines données expérimentales indiquent d’ailleurs que l’administration intramédullaire d’antagonistes de divers sous-types de récepteurs sérotoninergiques peut annuler l’effet antinociceptif du paracétamol.
L’action antithermique est liée à l’inhibition de la synthèse de PGE1 dans l’hypothalamus, l’organe responsable de la gestion de la thermorégulation.
5.2. Propriétés pharmacocinétiques
Méthocarbamol
5.2.1 Absorption
Après administration orale, il est absorbé de façon rapide et complète et atteint des concentrations plasmatiques maximales au bout de 1 à 3 heures. Les effets relaxants musculaires sont observables 30 minutes après l’administration de la dose orale.
5.2.2 Distribution
Une fois dans la circulation systémique, le méthocarbamol est largement distribué dans tout le corps. Chez des volontaires sains, sa liaison aux protéines plasmatiques est de 46 à 50 %. Chez les animaux de laboratoire, les concentrations les plus élevées sont détectées dans le foie et les reins. Ce médicament est capable de traverser la barrière placentaire, mais on ignore s’il est excrété dans le lait maternel.
5.2.3 Métabolisme
Le méthocarbamol est fortement métabolisé dans le foie par désalkylation et hydroxylation.
5.2.4 Élimination
Chez des volontaires sains, la clairance plasmatique du méthocarbamol oscille entre 0,2 et 0,8 l/h/kg, la demi-vie d’élimination étant de 1 à 2 heures. Le méthocarbamol est essentiellement éliminé dans l’urine, sous forme de métabolites glucuroconjugués et sulfoconjugués. Une petite quantité est excrétée dans les selles.
Patients âgés
Chez les patients âgés, la demi-vie d’élimination du méthocarbamol augmente légèrement par rapport aux sujets plus jeunes. D’autre part, la liaison aux protéines plasmatiques est quelque peu réduite (41 à 43 % contre 46 à 50 %).
Patients souffrant d’une insuffisance hépatique
Chez 8 patients souffrant d’une insuffisance hépatique due à une cirrhose alcoolique, la clairance totale du méthocarbamol a diminué d’environ 70 % par rapport à la population saine (11,9 l/h) et la demi-vie d’élimination a augmenté jusqu’à 3,38 heures (± 1,62) comparativement à 1,11 heure (± 0,27) chez les sujets sains. La fraction de méthocarbamol liée aux protéines plasmatiques a été de 40 à 50 %, inférieure à celle observée dans la population saine de même âge et de même poids (46 à 50 %).
Patients souffrant d’une insuffisance rénale
Chez les patients souffrant d’une insuffisance rénale, la clairance du méthocarbamol est également réduite.
Chez 8 patients souffrant d’une insuffisance rénale soumis à une hémodialyse d’entretien, la clairance du méthocarbamol a diminué de 40 % par rapport à la population saine, bien que la demi-vie d’élimination ait été similaire dans les deux groupes (1,2 heure contre 1,1 heure, respectivement).
Paracétamol
5.2.1 Absorption
Après administration par voie orale, la biodisponibilité du paracétamol est de 75 à 85 %. Il est absorbé largement et rapidement ; les concentrations plasmatiques maximales sont atteintes en un laps de temps qui varie de 0,5 à 2 heures en fonction de la forme pharmaceutique. Les médicaments qui retardent la vidange gastrique retardent également l’absorption du paracétamol.
5.2.2 Distribution
Le paracétamol absorbé se répartit uniformément dans tous les compartiments corporels, avec une concentration plus faible au niveau du tissu adipeux et du liquide céphalorachidien. Le degré de liaison aux protéines plasmatiques est d’environ 10 %. Il atteint son effet maximal au bout de 1 à 3 heures et la durée d’action est de 3 à 4 heures.
5.2.3 Métabolisme
Le métabolisme du paracétamol passe par une première étape hépatique dont la cinétique est linéaire. Toutefois, cette linéarité disparaît si les doses administrées sont supérieures à 2 g. Le paracétamol est essentiellement métabolisé dans le foie (90 à 95 %), plus de 90 % par conjugaison de son groupe hydroxyle avec l’acide glucuronique et des groupes sulfate. Toutefois, 5 % du paracétamol ingéré sont oxydés dans le foie par le cytochrome P450, conduisant à la formation de N-acétyl-para-benzoquinone-imine (NAPQI), métabolite responsable de l’hépatotoxicité. Aux doses thérapeutiques, la petite quantité de NAPQI produite est détoxifiée par conjugaison avec le glutathion réduit (donneur de groupes sulfhydryle). Lorsque les réserves de glutathion diminuent de 70 % ou plus, la NAPQI ne peut pas être détoxifiée, provoquant une nécrose centrolobulaire. Des doses élevées de paracétamol peuvent saturer les mécanismes habituels de métabolisation hépatique, ce qui entraîne l’utilisation de voies métaboliques alternatives produisant des métabolites hépatotoxiques et potentiellement néphrotoxiques par épuisement des réserves de glutathion.
5.2.4 Élimination
Le paracétamol est essentiellement éliminé dans l’urine sous forme conjuguée à l’acide glucuronique et, dans une moindre proportion, à des groupes sulfate ; l’excrétion sous forme inaltérée est inférieure à 5 %. Sa demi-vie d’élimination est de 1,5 à 3 heures (elle augmente en cas de surdosage, chez les patients souffrant d’une insuffisance hépatique, chez les personnes âgées et les enfants).
5.3. Données de sécurité précliniques
Paracétamol
Aux doses thérapeutiques, le paracétamol ne présente pas d’effets toxiques. Ce n’est qu’à des doses très élevées qu’il provoque une nécrose centrolobulaire hépatique chez l’animal et chez l’homme. De même, à très fortes doses, le paracétamol provoque une méthémoglobinémie et une hémolyse oxydative chez le chien et le chat, très rarement chez l’homme.
Chez le rat et la souris, des études de toxicité chronique, subchronique et aiguë ont montré des lésions gastro-intestinales, des anomalies de la numération sanguine, une dégénérescence du foie et du parenchyme rénal, voire une nécrose. Les causes de ces altérations ont été attribuées au mécanisme d’action d’une part et au métabolisme du paracétamol d’autre part. Chez l’homme, il semblerait également que les métabolites soient à l’origine des effets toxiques et des altérations correspondantes dans les organes. En outre, de très rares cas d’hépatite chronique agressive réversible ont été rapportés lors de l’utilisation prolongée (par ex. : 1 an) aux doses thérapeutiques. En cas de doses subtoxiques, des signes d’intoxication peuvent apparaître au bout de 3 semaines de traitement. Par conséquent, le paracétamol ne doit pas être administré à des doses élevées ni au long cours.
Des études supplémentaires n’ont pas mis en évidence de risque génotoxique significatif du paracétamol aux doses thérapeutiques, c’est-à-dire à des doses non toxiques.
Chez le rat et la souris, des études à long terme n’ont pas mis en évidence de tumeurs aux doses de paracétamol non hépatotoxiques.
Fertilité
Chez l’animal, les études sur la toxicité chronique démontrent que des doses élevées de paracétamol provoquent une atrophie testiculaire et une inhibition de la spermatogenèse ; on ignore l’importance de ces phénomènes chez l’homme.
Le paracétamol a montré un potentiel génotoxique et carcinogène (tumeurs du foie et de la vessie) chez le rat et la souris, associé à des doses hépatotoxiques. On considère néanmoins que cette activité génotoxique et carcinogène est liée à des modifications du métabolisme du paracétamol à des doses élevées et ne présente pas de risque pour son utilisation clinique.
Méthocarbamol
La toxicité aiguë du méthocarbamol est relativement réduite. Chez les animaux expérimentaux, l’intoxication se manifeste par une ataxie, une catalepsie, des crampes et un coma. Aucune étude de toxicité chronique n’a été réalisée. Les effets toxiques sur la reproduction n’ont pas été évalués. Il n’existe pas d’études à long terme chez l’animal pour évaluer le potentiel mutagène du produit. Aucune étude à long terme sur le potentiel carcinogène n’a été réalisée.
6 – DONNEES PHARMACEUTIQUES
6.1. Liste des excipients
Acide stéarique
Amidon de maïs prégélatinisé
Croscarmellose sodique
Stéarate de magnésium
Silice colloïdale anhydre
Povidone
Distéarate de glycéryle
Carboxyméthylamidon sodique de pomme de terre
Talc
6.2. Incompatibilités
Sans objet.
6.3. Durée de conservation
5 ans.
6.4. Précautions particulières de conservation
Conserver à une température inférieure à 30ºC.
6.5. Nature et contenu de l’emballage extérieur
Boîte de 50 comprimés sous plaquettes thermoformées (PVC/aluminium).
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières. Tout médicament non-utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
6.7. Inscription à une liste des substances vénéneuses
Liste II.
7 – TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
FAES FARMA, S.A.
Maxima Aguirre, 14 48940 Leioa
ESPAGNE
8 – NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
Enregistré à l’AEMPS sous le nº47.091
9. DATE DE PREMIERE AUTORISATION/RENOUVELLEMENT DE L’AUTORISATION
Date de première autorisation : 21-12-1968
Date du dernier renouvellement : décembre 2008
10 – DATE DE MISE A JOUR DU TEXTE
Novembre 2015
Dernière mise à jour de cette page
18/05/2020.
![]()